


Selective Group VII CO2 Reduction Electrocatalysts
Rhenium (Re) bipyridine-based catalysts are some of the most robust and well-characterized systems known for the kinetically selective proton-dependent reduction of carbon dioxide (CO2) to carbon monoxide (CO) and H2O. Using industrial Fischer-Tropsch technologies, CO can be converted into liquid fuels in the presence of hydrogen. The electrocatalytic production of liquid fuels could provide a route to carbon-neutral energy sources that would fit into the existing infrastructure, especially with regards to storage and transport.
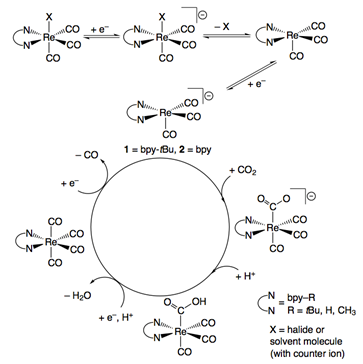
For these reasons our laboratory has undertaken significant investigations regarding these Re(I) bipyridine-based catalysts. Such studies have allowed us to develop a nuanced understanding of why these catalysts display a remarkable kinetic selectivity for the reduction of CO2, even in the presence of a large concentration of weak acid. A simplified mechanism for this catalyst family is shown below. The use of X-ray Absorption Spectroscopy to gain insight into the electronics of the active states of these catalysts and elucidation of the reaction mechanism using stopped-flow mixing in tandem with infrared or UV-Vis spectroscopy, has allowed us to begin exhaustive studies of the interaction of these catalysts with CO2.
Since rhenium is one of the least abundant elements in the Earth's crust, we have extended these studies to rhenium's first row Group VII counterpart, manganese. We have expanded on previous studies of manganese bipyridine catalysts by synthetically altering the bipyridine ligand to create more active catalysts. Significantly, these catalysts require the addition of weak acid for catalytic activity, whereas the rhenium catalysts are active in the absence of an external proton source. Through ongoing synthetic modification, however, we hope to achieve catalytic activities and stabilities that rival those of the rhenium bipyridine catalyst family. Our continued studies highlight the promise of the manganese catalyst family both for its ability to operate at a lower overpotential than its rhenium counterpart and for the earth-abundance of manganese.
The electrochemical reductive transformation of carbon dioxide is strongly hindered by the high-energy intermediates implicit in its direct reduction. We in the Kubiak lab are trying to work around these kinetic limitations by enabling concomitant proton and electron transfer in order to avoid these intermediates entirely. Certain biomimetic ligands, such as Lewis basic amines, can act as proton relays in the secondary-coordination spheres of catalysts for proton-dependent reductions. A series of catalysts containing these types of proton relays of particular note are the [Ni(P2N2)2]2+ complexes developed by Dr. Daniel DuBois. These complexes are functional hydrogenase mimics that have demonstrated some of the best electrocatalytic rates for hydrogen production and oxidation. Recently, our group has discovered that these complexes also have the fastest reported rate for the electrocatalytic oxidation of formate. Interestingly, the mechanism proposed for this transformation involves a proton-coupled 2-electron transfer event. As a part of our continuing studies we have developed new syntheses to greatly diversify the available P2N2 ligand catalog. By exploring the reactivity of different metals and novel ligands, we aim to adapt this proton-relay system for the reversible reduction of CO2.
Supported Molecular Catalysis
While homogeneous systems are favoured for their selectivity and tunability, heterogeneous catalysts have the advantages of stability, low catalyst loading, and straightforward
product separation. Linking molecular catalysts to conductive surfaces provides an opportunity to combine the advantages of
both systems. Our interest lies in finding the most optimum surface and attachment strategy for the molecular catalysts our group has studied over the past several
years. Factors such as attachment strength, orientation of the catalyst on the surface, stripping potentials, electron transfer, and catalyst loading are all areas
under investigation for these systems.
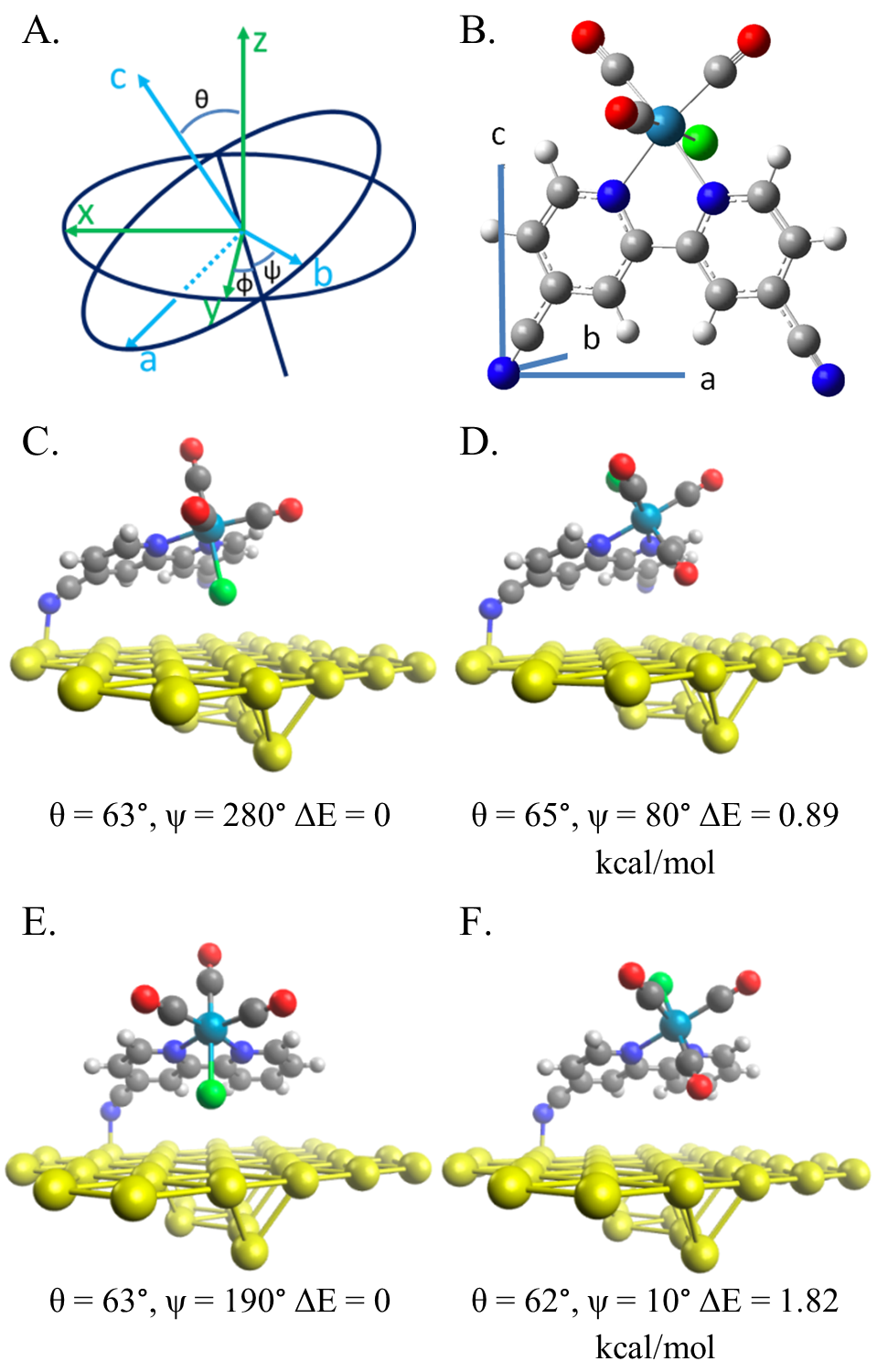
Tunable Catalysts Immobilized on Graphitic Supports
Another targeted strategy is to study heterogenized electrocatalyst/graphitic material hybrid electrodes. By understanding the electronic interactions between the carbon support
and the ligands of the electrocatalysts, we aim to synthetically control electronic coupling between catalyst and support and conjugate catalysts and supports into unique integrated
electrocatalyst materials. Catalyst design will combine control of substituents known to affect molecular catalysis and strong p-p coupling of catalyst and support. Graphitic materials,
such as carbon nanotubes, reduced graphene oxide, and carbon aerogels have been of interest to support electrocatalysts. Efforts have been focused on carbon nanotubes and carbon aerogels
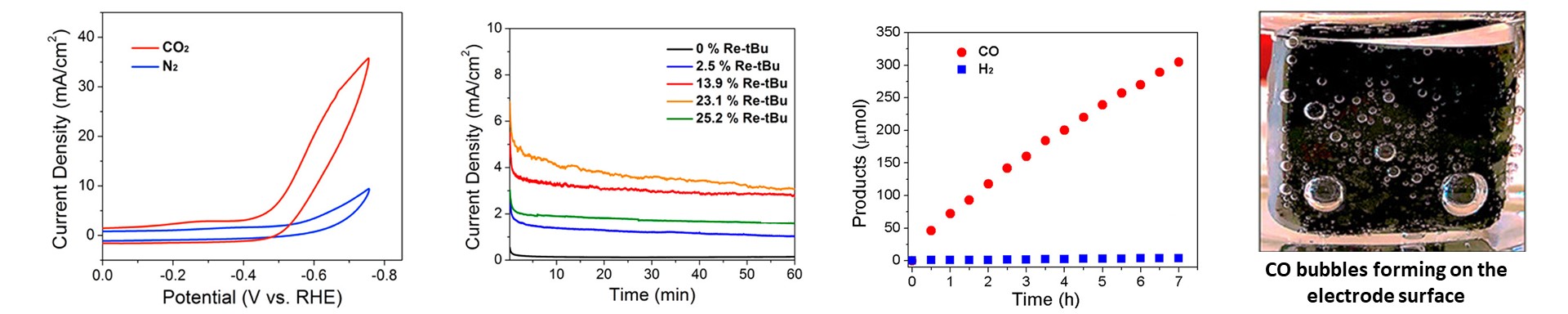
primarily. As shown in our recent work, Re(tBu-bpy)(CO)3Cl was immobilized onto multiwalled carbon nanotubes supported on glassy carbon. These hybrid electrodes were able to do CO2
reduction in near neutral pH aqueous electrolyte solutions. This hybrid system exhibited high selectivity for CO (near 100%), low overpotential, and high turnover frequency.
Further work is being conducted on quantifying and improving the percent of electroactive catalysts. Additionally, we are currently developing our electrode fabrication methods
by transitioning from drop-casting these hybrid electrodes onto glassy carbon to spray-coating onto carbon paper to make gas diffusion electrodes. This immobilization strategy allows
for direct comparisons of a variety of molecular catalysts in an aqueous environment. Also, comparisons can be made between a homogenous molecular catalyst and an immobilized molecular catalyst.
Investigation of Electron-Transfer Reactions in Highly Coupled Mixed-Valence Systems
Over the past 15 years our lab has studied electron-transfer reactions using the coalescence of ν(CO) band shapes in the IR spectra of mixed-valence dimer complexes of the type [Ru3O(κ2-OAc)6(CO)-(L)]2-BL, where L = an ancillary pyridyl ligand and BL = a cluster bridging pyrazine or 4,4'-bipyridine]. Based on these experiments we can estimate the rate constant of electron transfer (ET) present in singly reduced species. Thorough spectroscopic investigations of this system have allowed us to understand the environmental effects on ET dynamics occuring on the picosecond timescale. Studies of these mixed-valence complexes have provided a clear picture of ET dynamics at the Class II/III borderline1 of electron delocalization and have allowed for a more complete description of borderline-delocalized behavior.
Our most recent work involves the investigation of mixed valency in supramolecular systems and molecular electronics. Mixed valency across hydrogen bonds was described recently by our laboratory using these systems and is being investigated further to study the dynamics of proton-coupled ET reactions. In these hydrogen-bonded systems, electron delocalization appears to be a promising method to harden soft, noncovalent interactions. In addition to this, we have also recently published reports on nanoparticle- and quantum dot-based assemblies of mixed-valence redox-active metal centers. These reports detailed the exchange of electrons through both metallic and semiconducting systems with promising results toward generating systems that can achieve efficient photochemical charge separation. We have also begun to study photoinduced intramolecular electron transfer using porphyrins as coordinatively attached ancillary ligands. We hope to explore the dynamics involved as the dimer-of-trimer system evolves from a diabatic (uncoupled) state to an adiabatic (coupled) state, an extremely fast transition requiring a synthetically sophisticated system.
![]()
Demonstrations
Driving in Marcus Theory
The Marcus Theory of Electron Transfer describes the simplest chemical reaction: electron transfer of a single electron. The theory approximates the potential energy of the reactants and products as separate parabolas, and electron transfer is shown at their intersection. The x-axis is the reaction coordinate and the y-axis is the energy of the system. There are three main variables: ΔG* is the activation energy, ΔGr is the driving force (or free energy change), and λ is the reorganization energy.
There are three reactions shown, each with different potential energy surfaces. The first illustrates self-exchange, while the second and third depict bimolecular electron transfer reactions. The self-exchange reaction has no driving force, ΔGr, and therefore proceeds as quickly as the kinetic barriers allow. As the energy difference between the two potential energy surfaces increases, the ΔG* and ΔGr both increase and λ decreases. The consequence of this is that the rate of electron transfer is limited by the driving force and activation energy.
In the animation, the hills are formed from the parabolic potential energy surfaces of the reaction and the car represents the electron being exchanged. As the hill gets larger, the car slows down. This is analogous to Marcus Theory, as the rate of electron transfer slows as ΔG* and ΔGr increases.
Marcus Theory two and three state downloads for Matlab: two state three state
What do Chubby Checker and Berry Pseudorotation have in common?
1Described by the Robin-Day Classification of mixed-valence compounds; Robin, Melvin B.; Day, Peter. Mixed Valence Chemistry. Advances in Inorganic Chemistry and Radiochemistry. 1967, 10, 247-422.